Mediodia77
Plasta Culiad@
- Registrado
- 2005/09/17
- Mensajes
- 12.271
mentira weon, cientificos corruptos.
Follow along with the video below to see how to install our site as a web app on your home screen.
Nota: This feature currently requires accessing the site using the built-in Safari browser.

a la final el virus mutó de un murcielago o hubo intervencionyankeehumana?
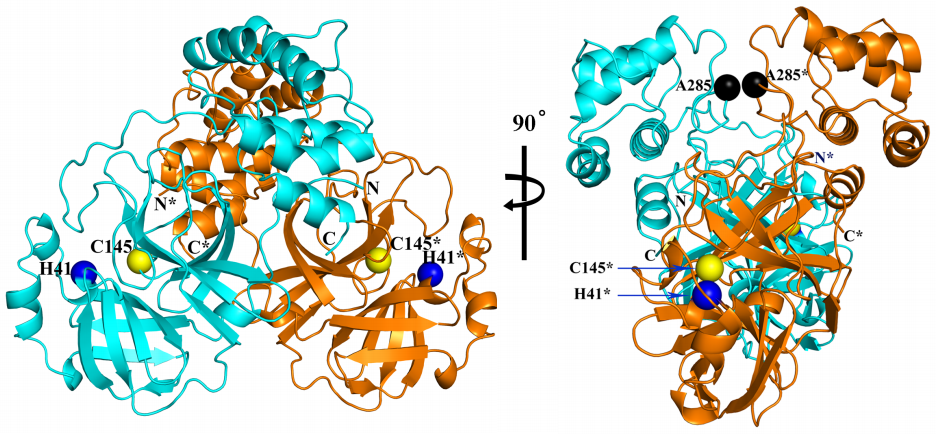
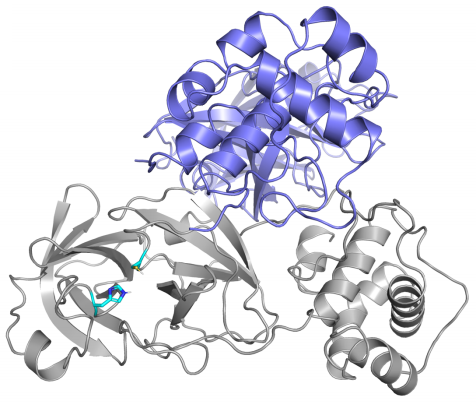
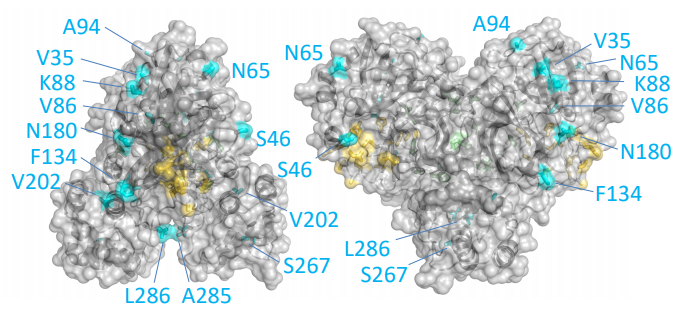
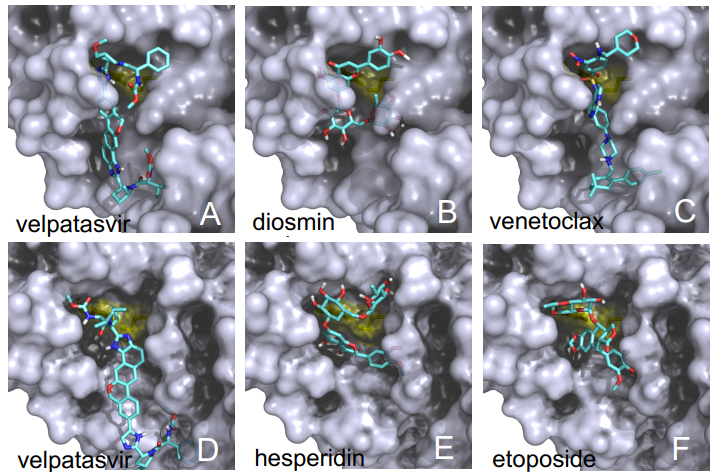
Las diferencias entre proteasa principal de SARS-CoV-2 y SARS-CoV son tan pequeñas que podemos confiar en la reconstrucción por homología de su estructura tridimensional. Gracias a ello podemos estudiar cómo interaccionan con esta proteasa diferentes antivirales, tanto los que atacan la cadena A (velpatasvir, diosmin y venetoclax) como los que atacan la cadena B (velpatasvir, hesperidin y etoposide). Por supuesto hay muchos más antivirales que inhiben esta proteasa, los de la figura son solo unos ejemplos representativos.
esa wea no era un monosacarido de los limones y naranjas (citricos)? o estoy confundiendo weas..? esto lo vi hace más de una década pero recuerdo que algo tenia que ver este tipo en a.v


No soy genetista, pero probablemente el número debe ser alrededor de los 100 para saber si existe realmente una diferencia significativa entre los genomas estudiados.Cuánto cantidad de datos es suficiente para determinar que hay nuevas variedades del virus ?
100, 200, 300, 1000 genomas ?
Hacían algo maravilloso conocido como secuenciación de Sanger. Escribiría una explicación, pero me di cuenta que tendría que escribir un mamotreto enorme para explicarlo bien, así que búscalo en youtube.Cómo lo hacían antes si no tenían la capacidad de secuenciación de hoy ?

No soy genetista, pero probablemente el número debe ser alrededor de los 100 para saber si existe realmente una diferencia significativa entre los genomas estudiados.
Hacían algo maravilloso conocido como secuenciación de Sanger. Escribiría una explicación, pero me di cuenta que tendría que escribir un mamotreto enorme para explicarlo bien, así que búscalo en youtube.
A la rápida, consiste en agregar di-desoxinucleótidos (ddNTPs) en una solución donde planeas hacer un PCR. Como los ddNTPs impiden que la reacción de la polimerasa se continúe, vas a terminar con un monton de fragmentos de distinto largo que, al ser sometidos a una electroforesis en agarosa (no recuerdo la concentración, probablemente sea como al 10%) se ordenarán por tamaño. La idea es hacerlo con un solo ddNTP en por reacción (ddATP, ddGTP, etc) para después poner el resultado de cada PCR en 4 bolsillos distintos. Como la electroforesis ordena los fragmentos por tamaño, vas a terminar con los fragmentos más chicos abajo y los más grandes arriba, entonces puedes leer la secuencia de tu fragmento de interés de abajo hacia arriba, de la siguiente forma:
Cómo lo hacían antes si no tenían la capacidad de secuenciación de hoy ?
 interesting .... por eso aún no se ponen de acuerdo qué animal fue, si el virus codifica diferentes proteínas similares a diferentes betacoronavirus en diferentes animales @.@
interesting .... por eso aún no se ponen de acuerdo qué animal fue, si el virus codifica diferentes proteínas similares a diferentes betacoronavirus en diferentes animales @.@

Depende de los datos mismos. Generalmente se considera cepas distintas cuando existe una cierta distancia genética entre un genoma y el otro. Después de todo no conviene denominar algo como una cepa o especie distinta si no existe ninguna diferencia funcional entre los dos genomas. En este caso se considera una subespecie probablemente porque una es más infecciosa que la otra.Opino similar a usted, creo que con 100 secuencias, si bien no es definitivo, es suficiente para mostrar una tendencia, más si la variedad nueva prevalece más que el ancestro (70% vs 30%) .... por qué no considerarlo como una nueva ?
Si no lo hace entonces es porque tu pregunta está mal formulada entonces. Se entiende que por "capacidad de secuenciación de hoy" te refieres a lo que es el Next Generation Sequencing, weas como Illumina, 434, etc. Se entiende "secuenciación masiva", weas con un throughput en las Gpb, no el campo completo de tecnologías de secuenciación. Antes del PCR moderno, creo que se secuenciaba en base a primers aleatorios que después se utilizaban para amplificar. Si amplificaba era porque la secuencia era correcta. Lo otro que se me puede ocurrir que hicieran sería digerir un ADN de interés con una nucleasa que sabes qué secuencia reconoce.Su respuesta no responde mi pregunta:
Es un aproximado más o menos exagerado, nunca he hecho un sanger personalmente, sólo RFLP, con el que ocupabamos agarosa al 2%. Probablemente sea algo más cercano al 5% que al 10%, lo exageré porque vas a necesitar ver los primeros nucleótidos de la secuenciación y con un gel del 2% se te van a caer o no los vas a distinguir (la resolución de un gel de 2% es de alrededor de las 200pb). El voltaje es en función de la distancia entre los electrodos, lo que aumentaría sería el amperaje.10% ? ? qué voltaje se debe utilizar para travesar todo ese gel super denso ? O:
Depende de los datos mismos. Generalmente se considera cepas distintas cuando existe una cierta distancia genética entre un genoma y el otro. Después de todo no conviene denominar algo como una cepa o especie distinta si no existe ninguna diferencia funcional entre los dos genomas. En este caso se considera una subespecie probablemente porque una es más infecciosa que la otra.
Si no lo hace entonces es porque tu pregunta está mal formulada entonces. Se entiende que por "capacidad de secuenciación de hoy" te refieres a lo que es el Next Generation Sequencing, weas como Illumina, 434, etc. Se entiende "secuenciación masiva", weas con un throughput en las Gpb, no el campo completo de tecnologías de secuenciación. Antes del PCR moderno, creo que se secuenciaba en base a primers aleatorios que después se utilizaban para amplificar. Si amplificaba era porque la secuencia era correcta. Lo otro que se me puede ocurrir que hicieran sería digerir un ADN de interés con una nucleasa que sabes qué secuencia reconoce.

TE GANASTE MIS RESPETOS, SAILOR MOONPodría leer el paper de Tang y dar su opinión ?
La mía es que está bien sus conclusiones, compara las mutaciones en sitios conservados en diferentes virus similares, cuenta con el cladograma respectivo, el ancestro tiene menos prevalencia que la mutación - eso no deja de ser curioso -, y para ambos sitios analizados cuentan con un LOD de 50,13. Quizá más bioinformática y secuencias podría ayudar a que el estudio sea más robusto, pero creo que más que eso, lo que falta es validar mediante análisis moleculares: evaluar expresiones de proteína que codifica ORF8 en células humanas de mutantes y el efecto de su combinatoria: fenotipeo, no es así como se identificaban nuevas variedades ?
Recordar al padre de la genética: Mendel