impedanci
El Marquez del Foraze
- Registrado
- 2008/10/04
- Mensajes
- 12.493
- Sexo

- MOH
Quizás has leído por ahí que hay dos linajes (o cepas) del coronavirus SARS-CoV-2, llamadas L y S, que solo se diferencian en un único aminoácido mutado; S sería ancestro de L y ésta se transmitiría más fácilmente (sería prevalente en el ~70% de los casos). En realidad, lo único que hacen los virus de ARN es replicarse y mutar; por ello toda persona infectada con el coronavirus SARS-CoV-2 tendrá en su interior genomas víricos con diferentes mutaciones. Entre el 29 de noviembre de 2019 y el 7 de marzo de 2020 se han secuenciado 209 genomas completos de SARS-CoV-2, en los que se han observado 111 mutaciones de aminoácidos no sinónimas (recuerda que el código genético es redundante y las mutaciones ocurren en los nucleótidos, luego hay muchas más mutaciones de nucleótidos). Ninguna de estas mutaciones de aminoácidos tiene relevancia clínica demostrada en la infección por COVID-19; luego no se puede afirmar que haya dos cepas (o más) bien separadas. Así que no te dejes engañar, por ahora solo existe una cuasiespecie de coronavirus SARS-CoV-2 entre la población humana.
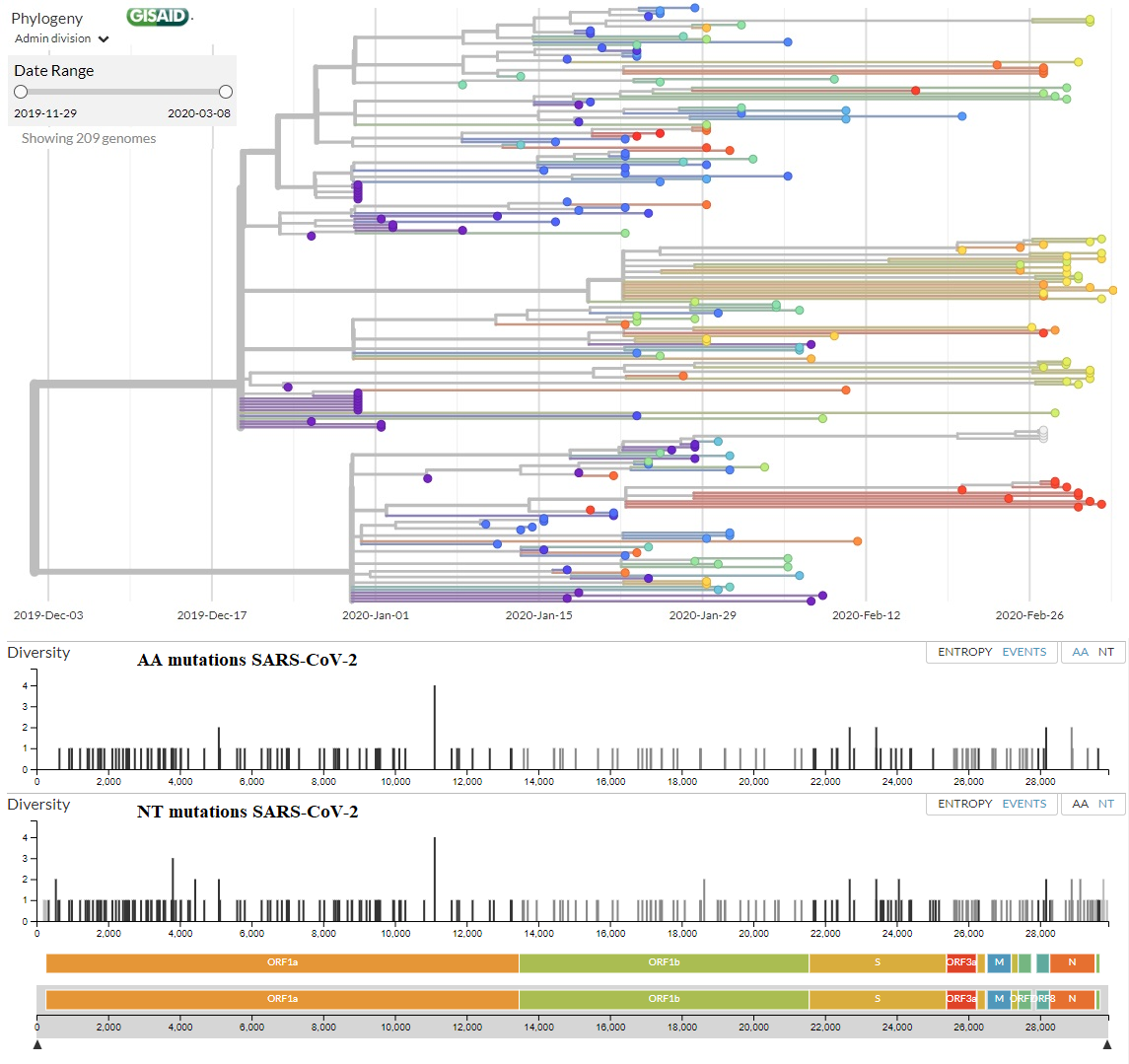
En esta figura te muestro el árbol filogenético (a fecha de 7 de marzo de 2020) con los 209 genomas secuenciados (la última versión actualizada la tienes en la web de NextStrain.Org, a partir de todos los genomas completos disponibles en la web de GISAID). En la parte inferior tienes todas las mutaciones de aminoácidos (AA mutations) y de nucleótidos (NT mutations) que se han encontrado, justo encima de los genes del coronavirus donde se han encontrado (puedes leer una explicación del genoma del SARS-CoV-2 en este blog en LCMF, 25 ene 2020). En el eje vertical tienes la diversidad de nucleótidos (Diversity) que mide el número promedio de diferencias entre nucleótidos en un sitio determinado entre dos secuencias ADN para todos los pares posibles en la población (más detalles y la fórmula que la calcula en la wikipedia). El listado de todas las mutaciones no sinónimas (de aminoácidos) lo tienes aquí, junto con el número de secuencias que la presentan.
Más información en Oscar A. MacLean, Richard Orton, …, David L. Robertson, “Response to “On the origin and continuing evolution of SARS-CoV-2”,” Virological.Org, 5 Mar 2020, quienes critican el artículo con la propuesta de las dos cepas de Xiaolu Tang, …, Jie Cui, Jian Lu, “On the origin and continuing evolution of SARS-CoV-2,” National Science Review, nwaa036 (03 Mar 2020), doi: https://doi.org/10.1093/nsr/nwaa036.
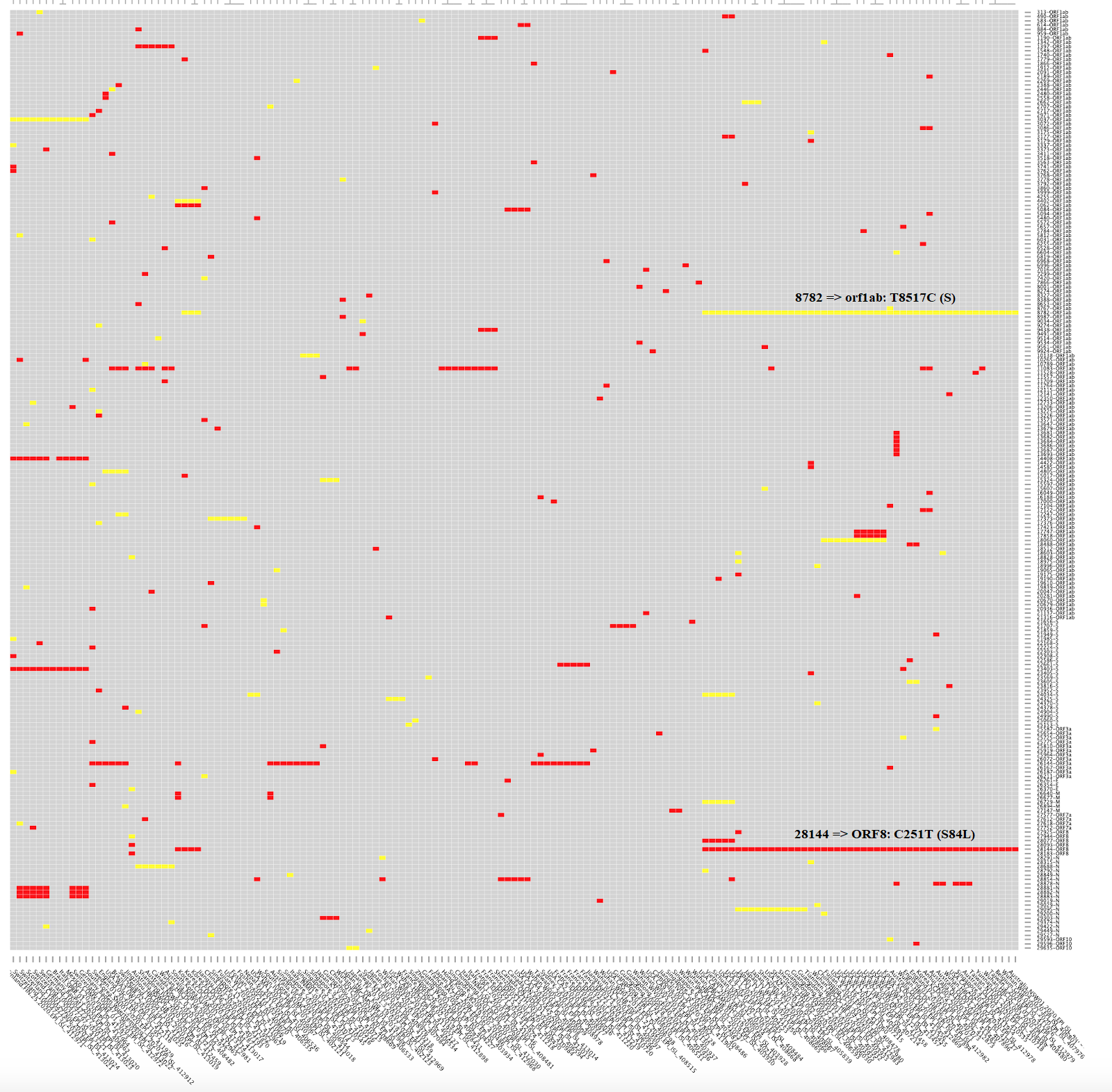
La crítica aún no publicada de MacLean et al. a la propuesta del artículo publicado por Tang et al. parece obvia para quien es consciente de que los coronavirus, como todos los virus de ARN, mutan mucho. Máxime cuando la propuesta es que la cepa S sea ancestro de la cepa L y que esta última se transmite más fácilmente (por ello, el ~70% de los infectados la tendría). Sin embargo, ambas cepas solo se diferenciarían en dos SNPs (se pronuncia esnips), polimorfismos de un solo nucleótido. En concreto, un SNP sinónimo en la posición 8782 del ARN, en el gen orf1ab, el cambio de una timina por una citosina en la posición 8517 de dicho gen (T8517C), que cambia el codón AGT de una serina por AGC también de una serina; y otro SNP no sinónimo en la posición 28144 del ARN, en el gen ORF8, el cambio de una citosina por una timina en la posición 251 de dicho gen (C251T), que cambia el codón de una serina por el de una leucina en la posición 84 (S84L). Esta figura muestra las 111 mutaciones no sinónimas en rojo y las restantes mutaciones sinónimas en amarillo; he aclarado dónde se encuentran las mutaciones 8782 (orf1ab: T8517C) y 28144 (ORF8: C251T, S84L).
La teoría evolutiva predice que el coronavirus SARS-CoV-2 está sometido a mutaciones continuas que producen una deriva genética que acabará conduciendo a su separación en diferentes cepas en el futuro. Lo habitual es que las cepas que mejor se adapten al ser humano (es decir, las que tengan una letalidad reducida y produzcan síntomas más leves) sean las que se acaben propagando con mayor facilidad. Pues al coronavirus le «interesa» sobrevivir el máximo tiempo posible entre los humanos y que no le pase lo que pasó con el SARS-CoV (cuya epidemia fue contenido y desapareció de la circulación entre los humanos).
Y, por cierto, seguro que en Twitter y en medios sensacionalistas has oído que el coronavirus SARS-CoV-2 podría haber escapado del laboratorio BSL-4 de Wuhan. Nada más lejos de la realidad. Se han publicado varios artículos que desmontan esta conspiración y demuestran que la variación genética natural de los betacoronavirus es suficiente para explicar la aparición de nuevos virus que puedan infectar a humanos (como ocurrió con SARS y MERS, y ahora con COVID-19). Lo que pasa es que las noticias virales como las del coronavirus acaban generando de manera espontánea gran número de conspiraciones, porque todos tenemos un poco de conspiranoicos aunque nos pese. Te recomiendo leer a Daniel Jolley, Pia Lamberty, El coronavirus es un campo abonado para los ‘conspiranoicos’,” The Conversation, 05 mar 2020.
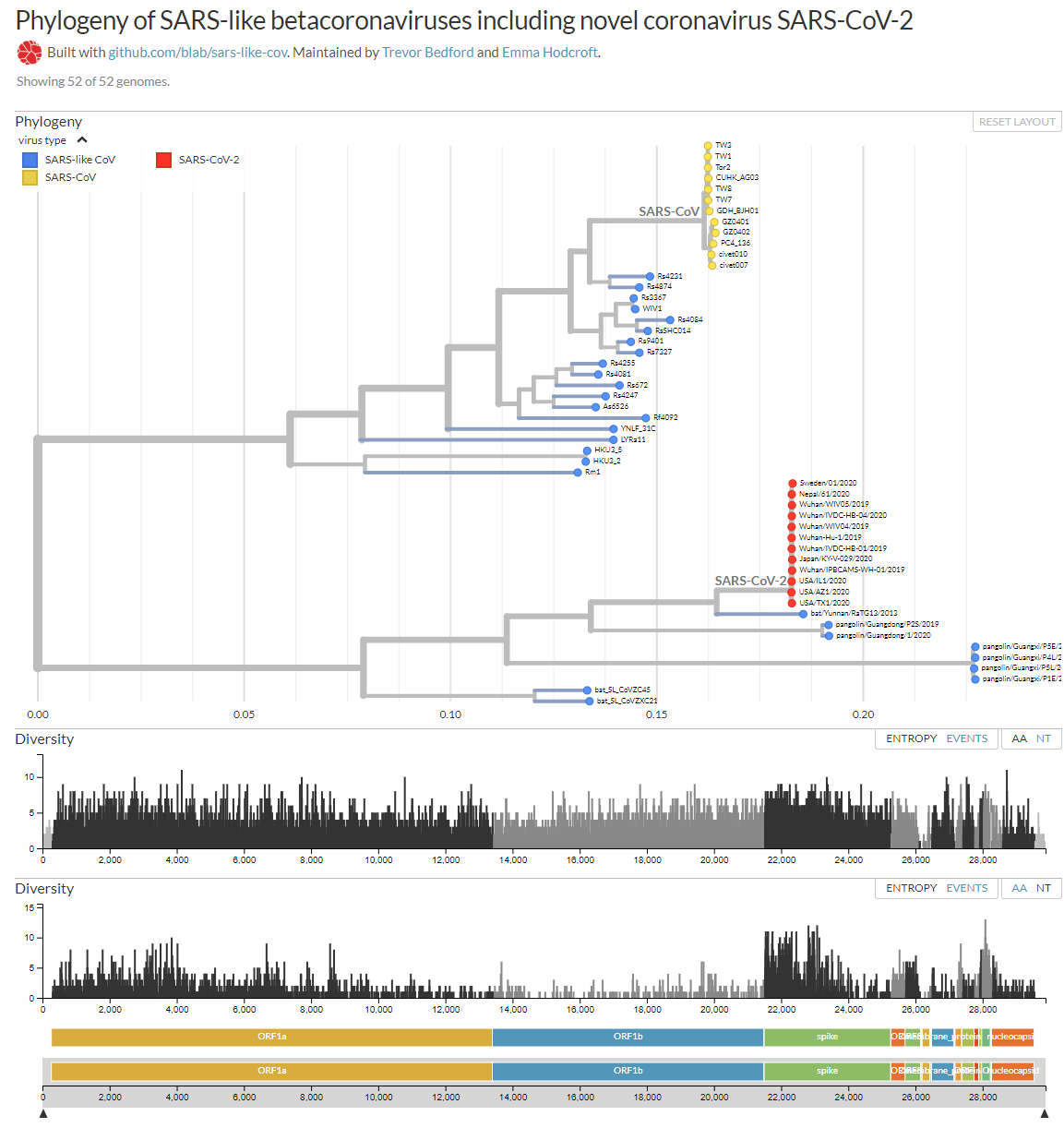
Me permito aportar mi granito de arena con esta figura que te muestra el arból filogenético de los betacoronavirus similares a SARS, entre ellos SARS-CoV-2, así como las mutaciones que se han observado en sus ARN. Prácticamente todos los nucleótidos (NT) han mutado alguna vez; y casi todos los aminoácidos (AA) también. Como se dispone de más genomas de los que infectan a humanos (SARS-CoV en amarillo y SARS-CoV-2 en rojo) que de los demás, se observa que la diversidad genética es mucho mayor en la glicoproteína S, responsable de la infección a humanos; pero su razón de ser es un simple sesgo estadístico. Como nos recuerda Ignacio López-Goñi en Twitter: “No, no se ha escapado de un laboratorio, ni es un arma biológica, la naturaleza se basta y se sobra para generar nuevos virus”.
En resumen, el coronavirus SARS-CoV-2 está mutando de forma continua como cuasiespecie que es, pero todavía no hay datos suficientes para afirmar que haya más de una cepa. Que no te engañen ni te asusten con supuestas mutaciones que incrementan su letalidad; lo más habitual es que las mutaciones más letales, si algún día llegan a aparecer, no se propaguen entre la población. Lo peor que nos puede pasar es que el virus se acomode a los humanos y se haga estacional como el virus de la gripe; en dicho caso tendremos epidemias de COVID-19 todos los años. Por fortuna, habrá vacunas y en poco tiempo todo el mundo se olvidará de su existencia (como han hecho con el virus de la gripe A). Eso sí, mucha gente en grupos de riesgo no se vacunará (como no se vacunan de la gripe) y fallecerá (como fallecen por la gripe), pero sus decesos ya no serán noticia en los medios.
https://francis.naukas.com/2020/03/07/las-mutaciones-del-coronavirus-sars-cov-2/
La estructura 3D de la glicoproteína espicular del coronavirus SARS-CoV-2

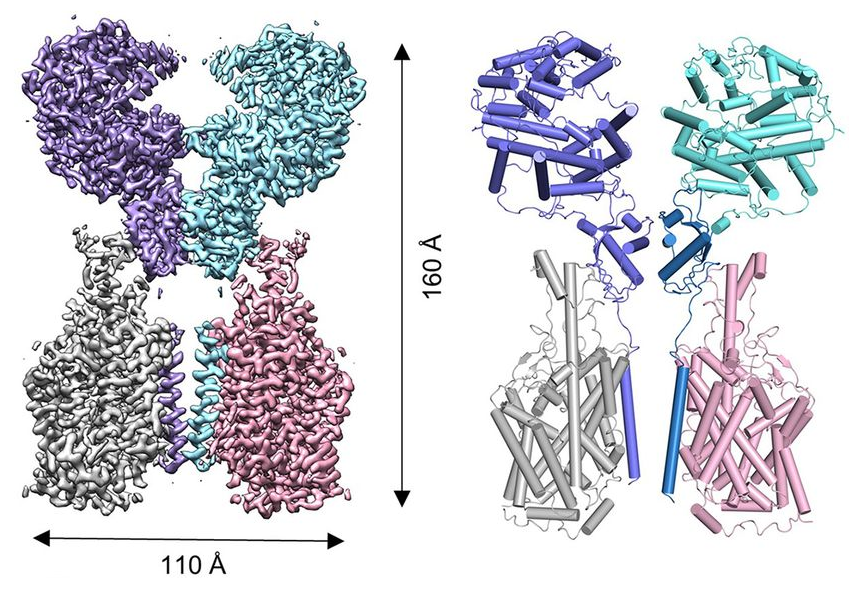
Desde que se publicó el genoma del betacoronavirus SARS-CoV-2 he deseado ver la imagen tridimensional de su glicoproteína espicular S. Como no podía ser de otra forma, su imagen con criomicroscopia electrónica se ha logrado en un tiempo récord. El equipo liderado por Jason S. McLellan, Univ. Texas en Austin (EEUU), la ha publicado en la revista Science. Gracias a esta imagen con una resolución de 3.5 Å se confirma que esta proteína S se acopla a la proteína ACE2 de las células humanas con mayor afinidad que la del coronavirus SARS-CoV. La proteína S es la diana de los anticuerpos que nos inmunizan. Su estructura 3D permite entender por qué los anticuerpos monoclonales publicados contra el SARS-CoV no son afectivos contra SARS-CoV-2. Sin lugar a dudas, ayudará a acelerar el desarrollo de vacunas y terapias contra la infección COVID-19.
La proteína S es un trímero formado por tres péptidos, cada uno con dos subunidades S1 y S2. La subunidad S1 actúa como una bisagra con dos conformaciones llamadas “abajo” (RBD down) y “arriba” (RBD up). La imagen por criomicroscopia electrónica muestra que solo uno de los péptidos está en estado “arriba”, estando los otros dos en estado “abajo”. La unión al receptor celular se realiza en la configuración “arriba”. Tras la unión se escinden los tres péptidos de la proteína S por el punto S1/S2; luego se produce una segunda escisión por el punto S2′, que despliega el péptido de fusión (FP) clave en la unión entre las membranas. La conformación tridimensional observada indica que la fusión entre el virus y el huésped es muy similar a la documentada en otros coronavirus (sobre todo para el coronavirus de la hepatitis murina, M-CoV, y para el SARS-CoV).
Sin lugar a dudas se ha dado un paso de gigante en la gestión de la infección COVID-19. El artículo es Daniel Wrapp, Nianshuang Wang, …, Jason S. McLellan, “Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation,” Science AOP eabb2507 (19 Feb 2020), doi: https://doi.org/10.1126/science.abb2507, bioRxiv preprint 944462 (15 Feb 2020), doi: https://www.biorxiv.org/content/10.1101/2020.02.11.944462. Por cierto, varios artículos han publicado reconstrucciones 3D realizadas por ordenador: Javier A. Jaimes, Nicole M. André, …, Gary R. Whittaker, “Structural modeling of 2019-novel coronavirus (nCoV) spike protein reveals a proteolytically-sensitive activation loop as a distinguishing feature compared to SARS-CoV and related SARS-like coronaviruses,” bioRxiv preprint 942185 (18 Feb 2020), doi: https://doi.org/10.1101/2020.02.10.942185; Alba Grifoni, John Sidney, …, Alessandro Sette, “Candidate targets for immune responses to 2019-Novel Coronavirus (nCoV): sequence homology- and bioinformatic-based predictions,” bioRxiv preprint 946087 (20 Feb 2020), doi: https://doi.org/10.1101/2020.02.12.946087; Chunyun Sun, Long Chen, …, Liangzhi Xie, “SARS-CoV-2 and SARS-CoV Spike-RBD Structure and Receptor Binding Comparison and Potential Implications on Neutralizing Antibody and Vaccine Development,” bioRxiv preprint 951723 (20 Feb 2020), doi: https://doi.org/10.1101/2020.02.16.951723; Renhong Yan, Yuanyuan Zhang, …, Qiang Zhou, “Structural basis for the recognition of the 2019-nCoV by human ACE2,” bioRxiv preprint 956946 (20 Feb 2020), doi: https://doi.org/10.1101/2020.02.19.956946; Jun Lan, Jiwan Ge, …, Xinquan Wang, “Crystal structure of the 2019-nCoV spike receptor-binding domain bound with the ACE2 receptor,” bioRxiv preprint 956235 (20 Feb 2020), doi: https://doi.org/10.1101/2020.02.19.956235; también se ha reconstruido la estructura de la proteasa M del coronavirus en Linlin Zhang, Daizong Lin, …, Rolf Hilgenfeld, “X-ray Structure of Main Protease of the Novel Coronavirus SARS-CoV-2 Enables Design of α-Ketoamide Inhibitors,” bioRxiv preprint 952879 (20 Feb 2020), doi: https://doi.org/10.1101/2020.02.17.952879.
[PS 25 feb 2020] Se ha publicado el preprint de una segunda reconstrucción 3D de la proteína S del SARS-CoV-2 obtenida mediante criomicroscopia electrónica, obtenida por el equipo de David Veesler (Univ. Washington, Seattle, USA). Un artículo muy interesante que complementa al publicado en Science. Los interesados disfrutarán con Alexandra C. Walls, Young-Jun Park, …, David Veesler, “Structure, function and antigenicity of the SARS-CoV-2 spike glycoprotein,” bioRxiv preprint 956581 (20 Feb 2020), doi: https://doi.org/10.1101/2020.02.19.956581. [/PS]
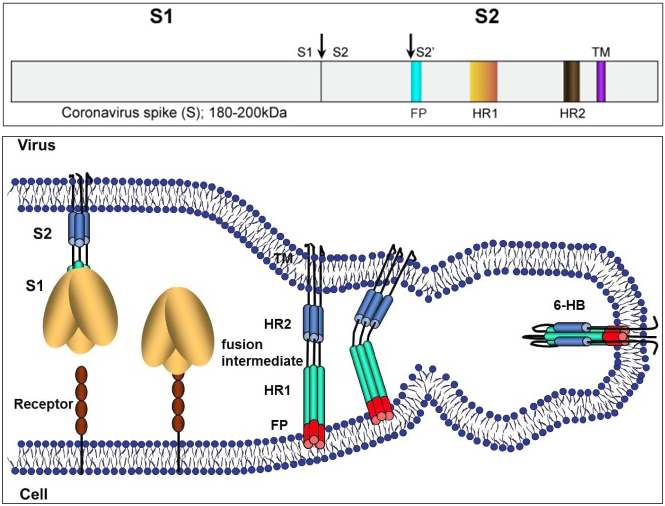
Fuente: Spike Protein / S Protein (Sino Biological).
La proteína S (espicular) es una proteína trimérica transmembrana de tipo I con entre 1160 y 1400 aminoácidos, según el tipo de coronavirus. Esta proteína que forma la «corona» de los coronavirus está constituida por tres pépticos repetidos (es un trímero) y está muy glicosilada (contiene entre 21 y 35 sitios donde se adicionan carbohidratos), lo que facilita su unión a azúcares y proteínas. Cada péptido está formado por dos dominios llamados S1 y S2. En los gammacoronavirus y en algunos betacoronavirus se produce una escisión de las subunidades S1 y S2 durante la fusión entre las membranas, mientras que en los alfacoronavirus y en algunos betacoronavirus no se produce dicha escisión. SARS-CoV-2 es un betacoronavirus en el que se produce la escisión.
El dominio S1 tiene a su vez dos subdominios, uno N-terminal (NTD), que finaliza con un aminoácido que posee un grupo amino libre (-NH2), y otro C-terminal (CTD), que termina en un grupo carboxilo (-COOH); ambos se acoplan al receptor ACE2 de la célula huésped, luego son dominios de unión al receptor (RBD). El dominio S2 es de tipo C-terminal y está muy conservado entre todos los coronavirus, que se diferencian mucho más en la subunidad S1. El dominio S2 contiene dos regiones, HR1 y HR2, en las que se repiten grupos de siete aminoácidos (llamados heptads), en la forma abcdefg, conteniendo a y d residuos hidrófugos que participan en la fusión entre las membranas. Los dominios HR1 y HR2 son dianas terapéuticas, pues se conocen fármacos que inhiben su acción, evitando o dificultando la fusión.
La infección de células epiteliales de las vías respiratorias está orquestada por la proteína S del virus. La figura ilustra los pasos generales del proceso de fusión. Primero, el dominio S1 reconoce y se acopla al receptor de la célula huésped. Segundo, se produce una primera escisión de los dominios S1 y S2, y una segunda escisión en el punto S2′; esta última permite que se active el péptido de fusión (FP) que conecta las membranas del huésped y el virus (esta fase se llama etapa intermedia de fusión, o fusion-intermediate stage). Y tercero, la región entre HR1 y HR2 se reconforma (se dobla) dando lugar a un heptámero (6-HB) que une ambas membranas permitiendo la entrada del virus.
La proteína S de los coronavirus es clave en el desarrollo de vacunas (antígenos que induzcan una respuesta inmune a la presencia del dominio S1) y para el desarrollo de antivirales (inhibidores de algunas de las etapas de la fusión entre membranas, normalmente atacando regiones concretas del dominio S2). Conocer la estructura tridimensional de la proteína S es fundamental para combatir la epidemia de COVID-19.
Este vídeo del laboratorio de David Veesler, Univ. Washington (EEUU), muestra la glicoproteína S del coronavirus más conocido y estudiado, el virus de la hepatitis murina (MHV), que infecta al ratón. Se destacan en el vídeo el gran número de regiones glicosiladas (los azúcares que decoran a la proteína) que ayudan al virus a evadir el reconocimiento por parte de anticuerpos. Supongo que en las próximas semanas se publicará un vídeo similar con la del coronavirus SARS-CoV-2. Por cierto, se publicó en Alexandra C. Walls, M. Alejandra Tortorici, …, David Veesler, “Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer,” Nature 531: 114-117 (08 Feb 2016), doi: https://doi.org/10.1038/nature16988.
El nuevo artículo en Science incluye un vídeo que muestra cómo cambia la conformación de la glicoproteína espicular S del coronavirus SARS-CoV-2 antes y después de fusionarse al receptor ACE2 (segunda enzima convertidora de angiotensina I) en la membrana de la célula huésped. Se observa tanto en una vista frontal, al principio del vídeo, como en una vista superior, al final. En el paso de la subunidad S1 del estado “abajo” (RBD down) al estado “arriba” (RBD up) se observa también un cambio de la conformación de la subunidad S2.

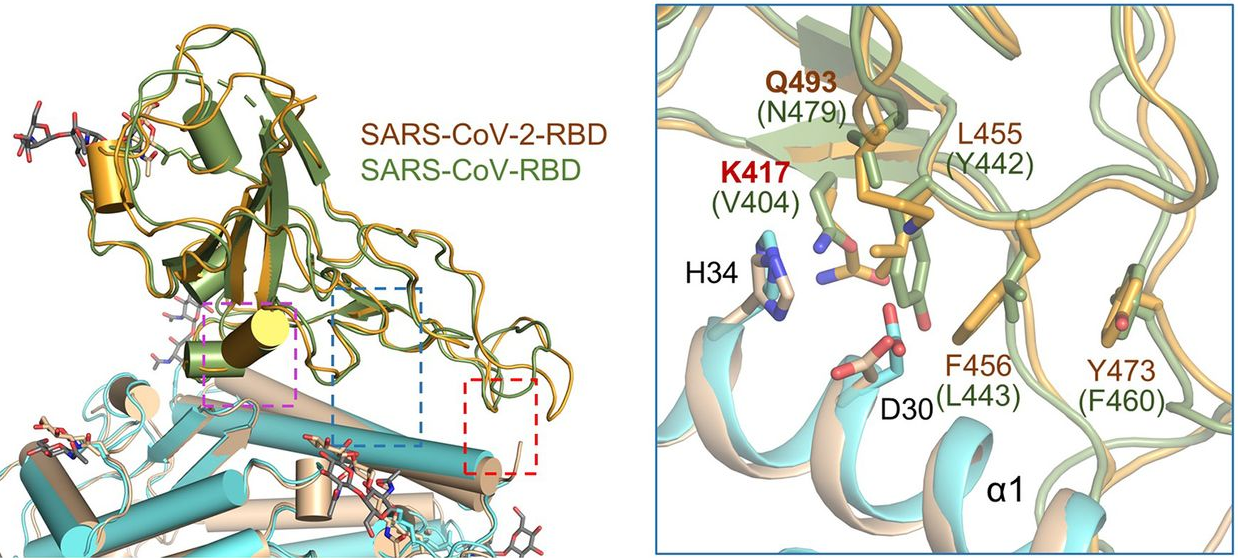
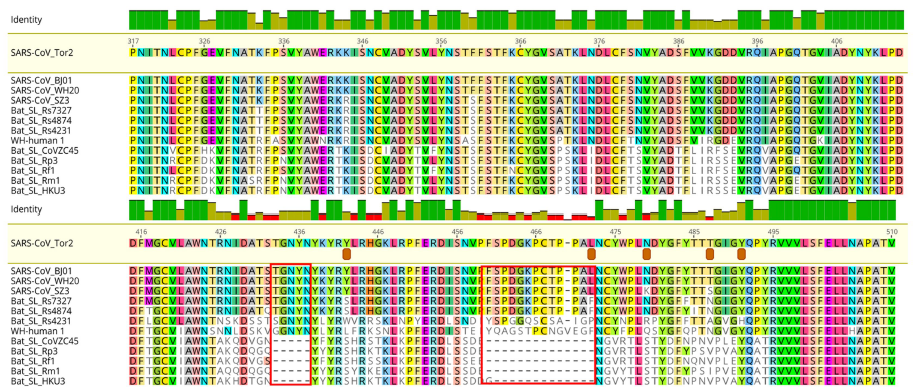
La secuencia de la proteína S del SARS-CoV-2 coincide al 98% con la proteína S del coronavirus Bat-RaTG13, con la gran diferencia de que en el sitio de escisión se encuentran los cuatro aminoácidos RRAR (arginina-arginina-alanina-arginina) en lugar de una sola arginina (R). Además, se diferencian en 29 residuos, 17 de los cuales se encuentran en la región RBD. La comparación realizada en el artículo entre los 61 genomas completos de SARS-CoV-2 disponibles en GISAID (Global Initiative on Sharing All Influenza Data) muestra que solo 9 aminoácidos diferentes entre todos ellos; y todas estas variantes están en lugares muy bien conservados, con lo que no parecen afectar a la letalidad del coronavirus.
En resumen, un artículo muy interesante que tendrá un gran impacto entre quienes luchan contra la infección de COVID-19. En bioquímica se dice que la forma determina la función. Poder observar la estructura tridimensional de la proteína S permite explorar con nuevos ojos los detalles de los procesos bioquímicos involucrados en la fusión entre el virus y la célula huésped.





 Lo peor que nos puede pasar es que el virus
Lo peor que nos puede pasar es que el virus